ARCHEOLOG-HOME / INDIana-UNIversitas
Contact























Global diversity, population stratification, and selection of human copy-number variation
Source - http://www.sciencemag.org/content/349/6253/aab3761.abstract
INTRODUCTION
Most studies of human genetic variation have focused on single-nucleotide variants (SNVs). However, copy-number variants (CNVs) affect more base pairs of DNA among humans, and yet our understanding of CNV diversity among human populations is limited.
RATIONALE
We aimed to understand the pattern, selection, and diversity of copy-number variation by analyzing deeply sequenced genomes representing the diversity of all humans. We compared the selective constraints of deletions versus duplications to understand population stratification in the context of the ancestral human genome and to assess differences in CNV load between African and non-African populations.
RESULTS
We sequenced 236 individual genomes from 125 distinct human populations and identified 14,467 autosomal CNVs and 545 X-linked CNVs with a sequence read-depth approach. Deletions exhibit stronger selective pressure and are better phylogenetic markers of population relationships than duplication polymorphisms. We identified 1036 population-stratified copy-number–variable regions, 295 of which intersect coding regions and 199 of which exhibit extreme signatures of differentiation. Duplicated loci were 1.8-fold more likely to be stratified than deletions but were poorly correlated with flanking genetic diversity. Among these, we highlight a duplication polymorphism restricted to modern Oceanic populations yet also present in the genome of the archaic Denisova hominin. This 225–kilo–base pair (kbp) duplication includes two microRNA genes and is almost fixed among human Papuan-Bougainville genomes.
The data allowed us to reconstruct the ancestral human genome and create a more accurate evolutionary framework for the gain and loss of sequences during human evolution. We identified 571 loci that segregate in the human population and another 2026 loci of fixed-copy 2 in all human genomes but absent from the reference genome. The total deletion and duplication load between African and non-African population groups showed no difference after we account for ancestral sequences missing from the human reference. However, we did observe that the relative number of base pairs affected by CNVs compared to single-nucleotide polymorphisms is higher among non-Africans than Africans.
CONCLUSION
Deletions, duplications, and CNVs have shaped, to different extents, the genetic diversity of human populations by the combined forces of mutation, selection, and demography.
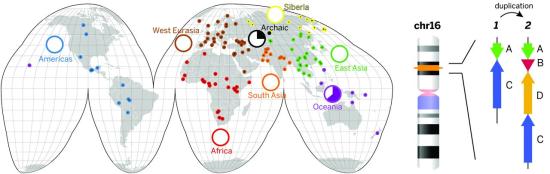 Figure Global human CNV diversity and archaic introgression of a chromosome 16 duplication.
Figure Global human CNV diversity and archaic introgression of a chromosome 16 duplication.
(Left) The geographic coordinates of populations sampled are indicated on a world map (colored dots). The pie charts show the continental population allele frequency of a single ~225-kbp duplication polymorphism found exclusively among Oceanic populations and an archaic Denisova. (Right) The ancestral structure of this duplication locus (1) and the Denisova duplication structure (2) are shown in relation to their position on chromosome 16. We estimate that the duplication emerged ~440 thousand years ago (ka) in the Denisova and then introgressed into ancestral Papuan populations ~40 ka.